SolX is a coordinate-based solution X-ray scattering simulation computer program. It is a stand-alone, Windows-based software. The installation package can be downloaded here.
SolX Graphic User Interfaces
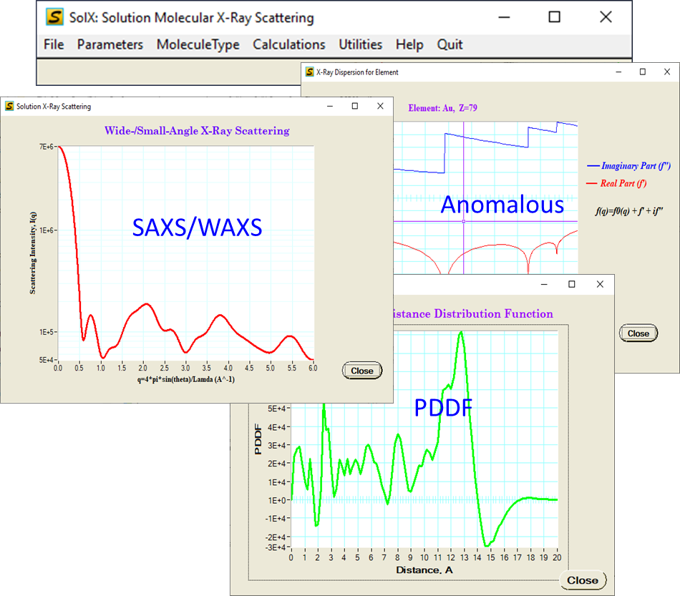
Main simulation functions:
1. Molecular X-ray Scattering
2. Molecular Pair Distance Distribution Function (PDDF)
3. Molecular Anomalous X-ray Scattering and PDDF
4. Reduced Molecular Structure Function and PDF/G(r)
Any questions/comments/sugguestion, please contact:
Xiaobing Zuo: zuox(AT)anl.gov
David Tiede: tiede(AT)anl.gov
A. X-ray Scattering Calculation
SolX program computes the scattering profile using Debye formula:
`A_(j)(q) = f_{j}(q)e^{cB_{j}q^2/16\pi} -g_{j}(q)` -- Eqn (2)
`f_{j}(q)` is the form factor of jth atom; `r_{j,k}` is the distance between jth and kth atoms; and `g_{j}(q)` is the dummy atom form factor
B. Coordinate-based pair distance distribution function
Pair distance distribution function (PDDF, P(r)) is also calculated from structural coordinates. P(r) and I(q) are related by Equantion (3):
`P(r) = frac(1)(4\pi^3)int_0^{\infty}I(q)*r*q*sin(r*q)dq ` -- Eqn (3).
Since `A_j(q)` is a summation of Gaussians, substitute Eqn (1,2) into Eqn(3), `P(r)` can be written as a summation of a series of Gaussian functions in terms of (`r` and `r_{j,k}`). .
C. Anomalous X-ray Scattering
When the incident X-ray energy is in the vicinity of the atomic X-ray absorption edge, the atomic form factor(`f(q)`) in Eqn (1,2) needs an anomalous dispersion correction :
`f_j^E(q,E) = f_j^0(q) + f_j^'(E) + i f_j''(E) ` -- Eqn (4).
`f_j^0(q)` is the normal atomic form factor in Eqn (2) and x-ray energy `E`-independent, but `q`-dependent. `f_j^'` and `f_j''` are real and imaginary correction, both are `E`-dependent, but `q`-independent. In anomalous calculations, `f_j^E(q,E)` will be used in Eqn 1-3, instead of `f_j^0(q)`.
SolX Short Instruction
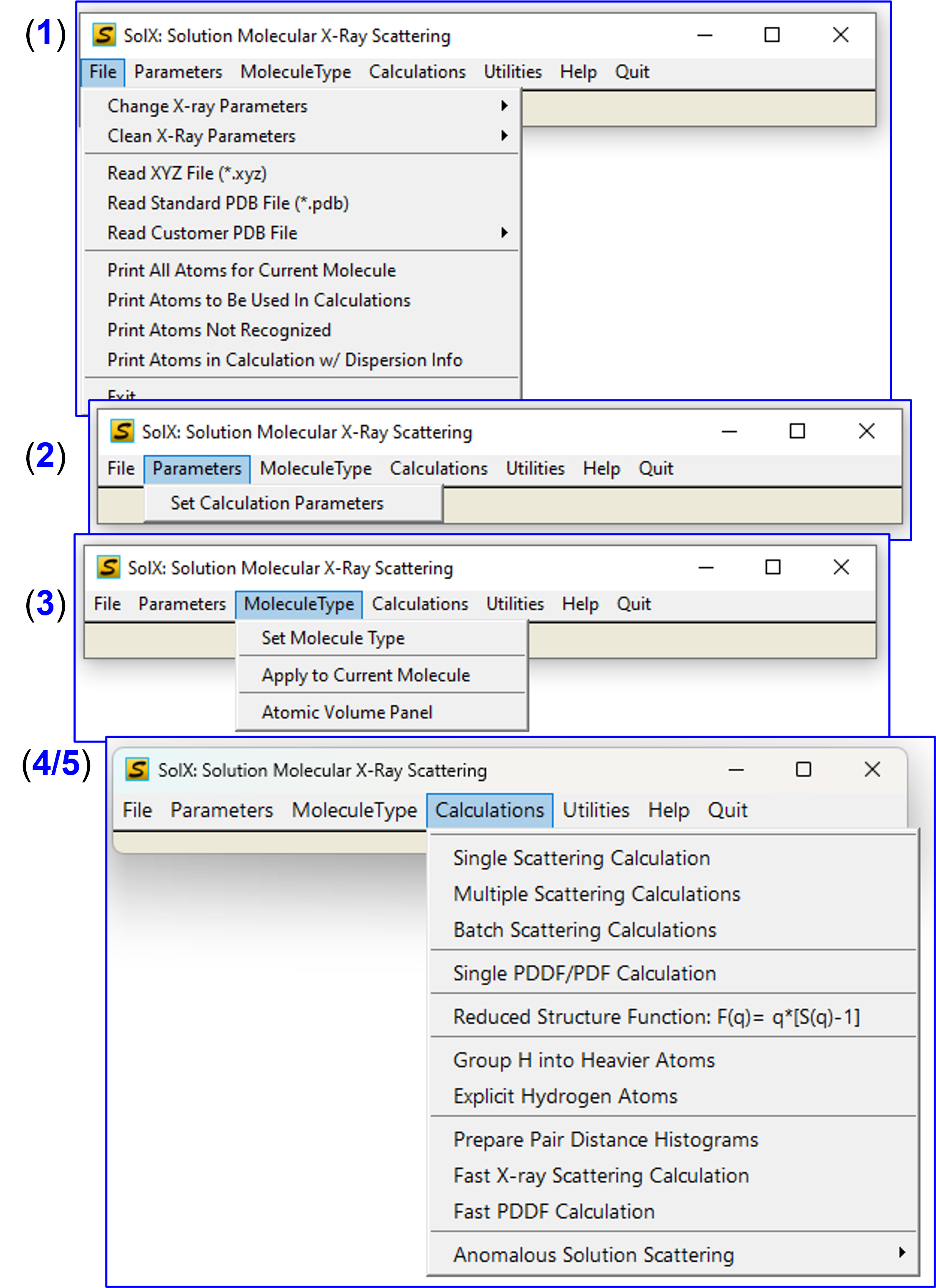
SolX Menu GUI
"Fast X-ray Scattering Calculation" and "Fast PDDF Calculation" are recommended for large molecules(~>50 kDa). They use pair distance histogram algorithm.
In addition to X-ray scattering, there are other types of calculations available under this menu, for example PDDF/PDF and anomalous etc. More details on these calcuations in later sections.
0. SolX menu GUI
SolX Menu GUI
Sub menu: File, Parameters, MoleculeType, Calculations, Utilities
1. File sub-menu
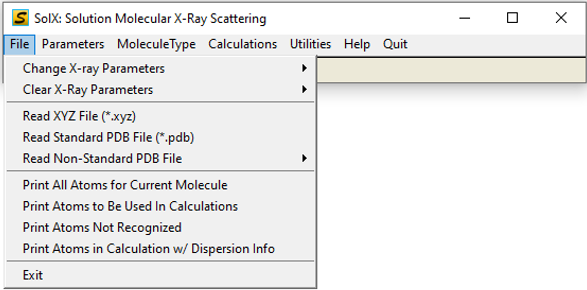
File Menu GUI
2. Parameters sub-menu
SolX Menu GUI
SolX Menu GUI
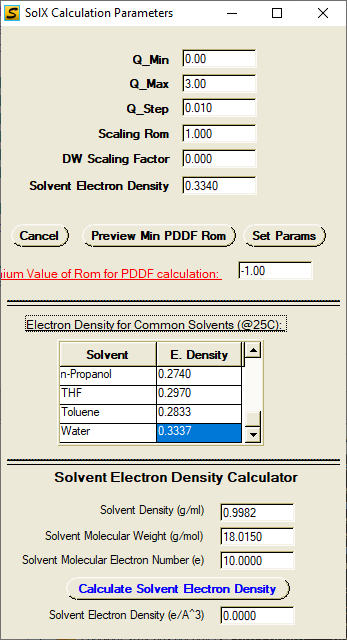
Set parameters for X-ray scattering (including anomalous) calculations: Q_Min, Q_Max, Q_step, Scaling Rom, DW Scaling Factor, Solvent Electron Density.
3. MoleculeType sub-menu
MoleculeType sub-menu
SolX Menu GUI
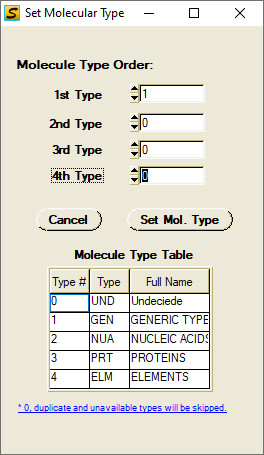
Set Molecule Type and Modify Atomic Volume Parameters if needed.
4. Calculations sub-menu
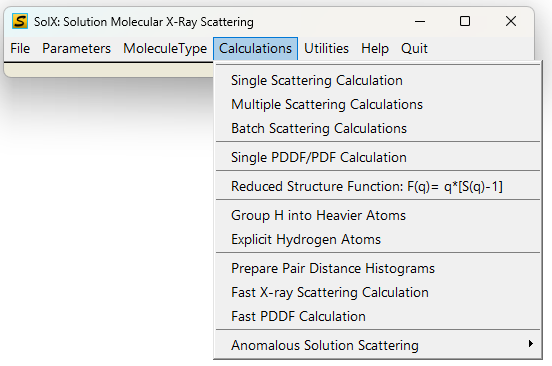
Calculations Menu GUI
SolX Calculations include:
A. Scattering Calculations
Single Scattering Calculation
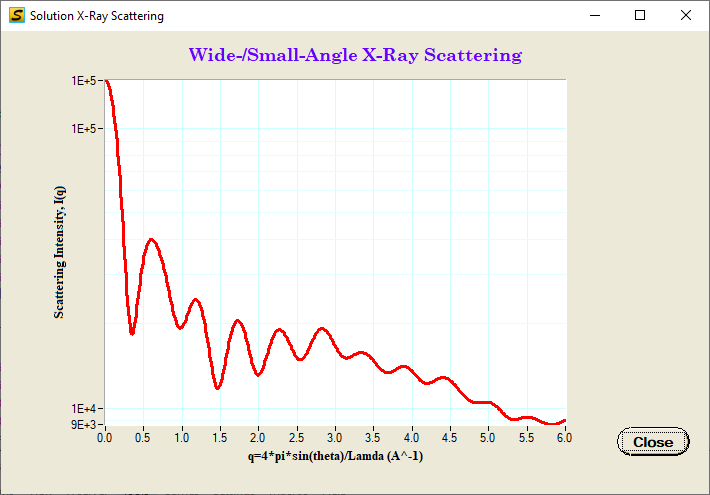
B. PDDF & G(r) Calculations
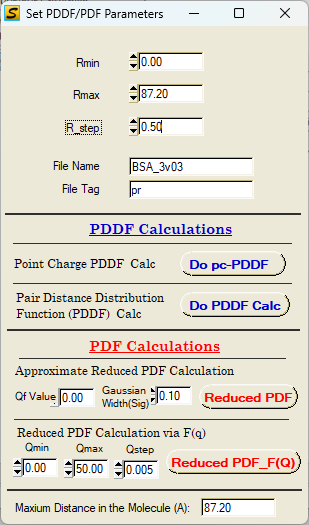
Single PDDF/G(r) Calculation
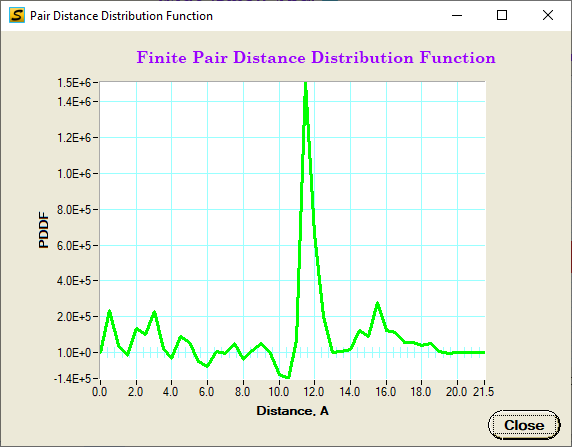
PDDF and reduced PDF calculation:
C. Anomalous X-ray Scattering Calculations
Anomalous Calculation Menu
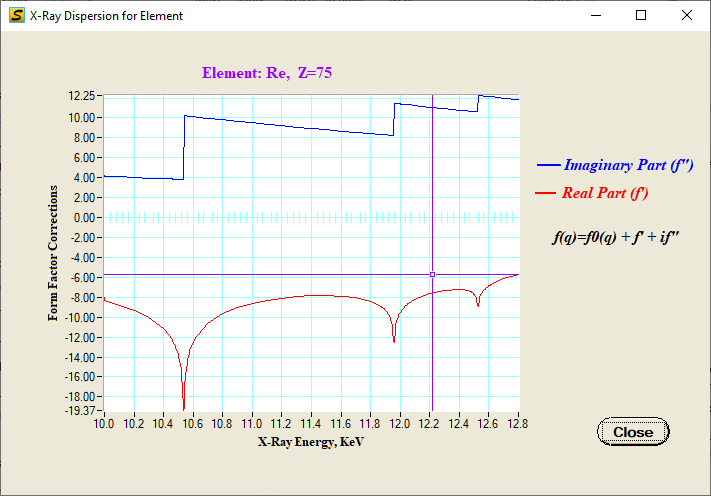
Display dispersion
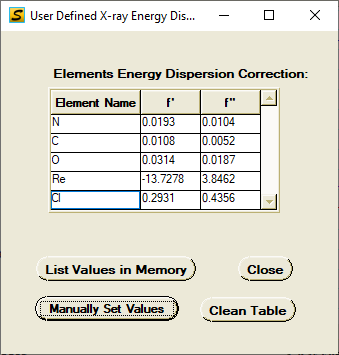
X-ray dispersion values
Anomalous X-ray Scattering Calculation:
D. Reduced Structure Function F(q)=q[S(q)-1]
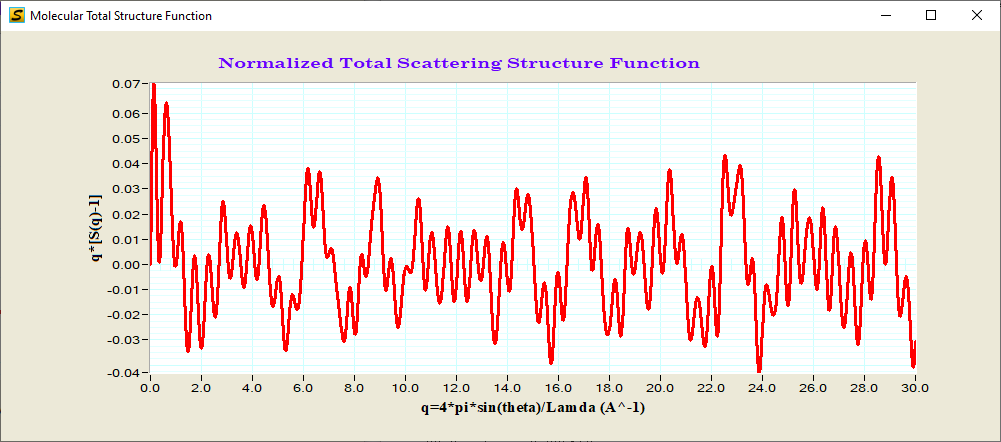
Reduced Structure Function
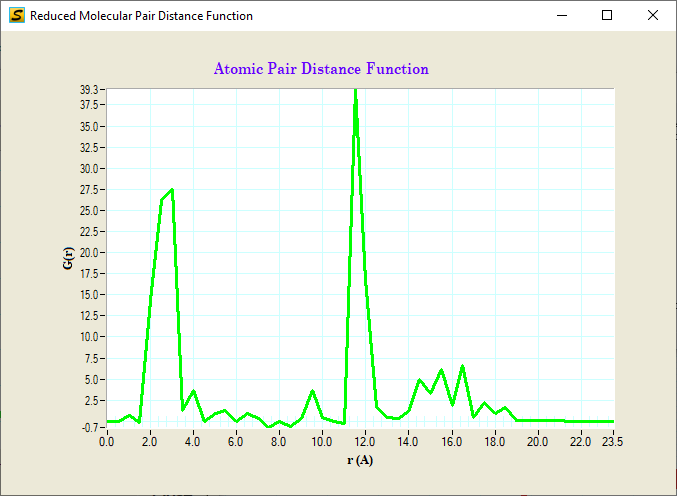
G(r) or reduced PDF
Reduced structure function and PDF:
5. Utilities sub-menu
Utilities Menu GUI
SolX Utilities include:
User can define new parameter values, such as new atom types, new atom names, new volume values, new form factors, or overwrite existing
parameters in built-in library.
Those can be achieved by editing three text files under SolX program folder:
SolXAtoms.txt, SolXAtomMap.txt and SolXFormFactor.txt.
Parameters defined in those .txt-files will be loaded along with those built-ins,
but will over-write existing built-in parameters.
1. SolX Atom Type
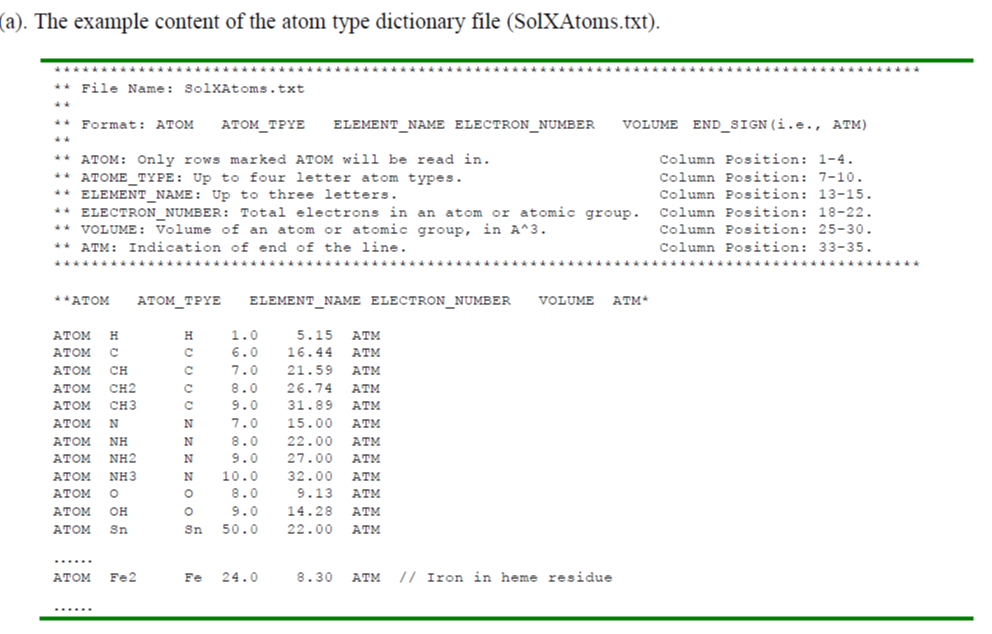
SolX Atome Type format
SolX Atom Type can be defined in SolXAtoms.txt, with following format
2. Atom Name Conversion Map

SolXAtomMap format
Current SolX program is a 32-bit version. Minimum Operating System requirement is Windows XP. The installation is straightforward,
simply following the on-screen instruction and clicking "Next>>", except for changing "Target directory for application" as shown below.
If the program behaves abnormally, uninstall and reinstall it.
A. Installation
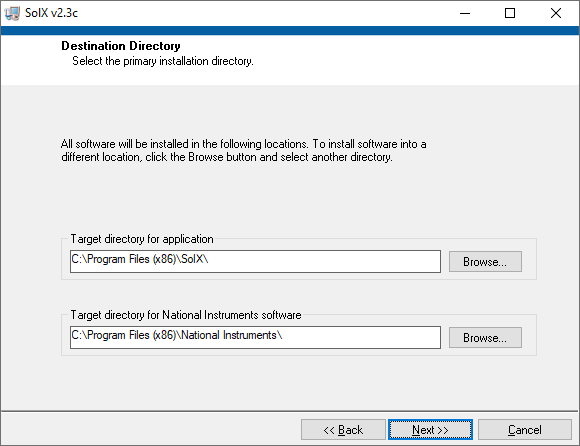
SolX installation 1
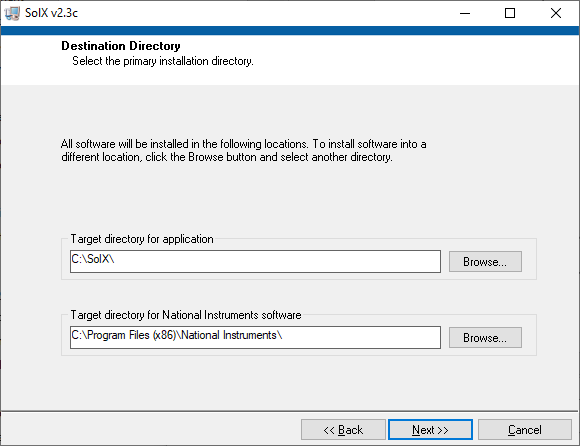
SolX installation 2: Change "Target directory for application"
SolX installation 3
Quick instruction on installation
B. Uninstallation
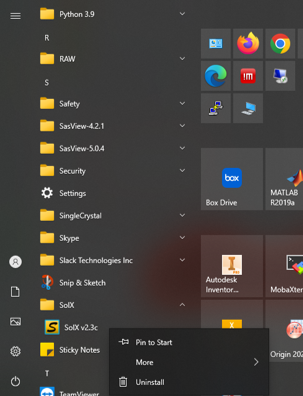
Starting uninstallation from "Start" menu
Uninstall at "Control Panel"--> "Programs"
Examples and Publications using SolX:
1. Examples

beta-cyclodextrin